Prepare 3 tab format for RNAseq dataset uploading
The following R code goes through preparing a dataset for upload to gEAR using the 3 tab format. If you wish for assistance with the formatting or upload process, please contact our curator team (https://umgear.org/contact.html)
- load in count file
countsFileName="count.txt"
count = read.delim(sep="\t",countsFileName, header=T, check.names=F)
names(count)[1] = "gene"
rownames(count) = count$gene
- generate the observation file (this can be done either via code or creating a tab delimited file in Excel.
options(repr.matrix.max.rows=50, repr.matrix.max.cols=200)
meta = read.delim("phenotype.txt")
#re-name the columns, change gender to your variable of interest
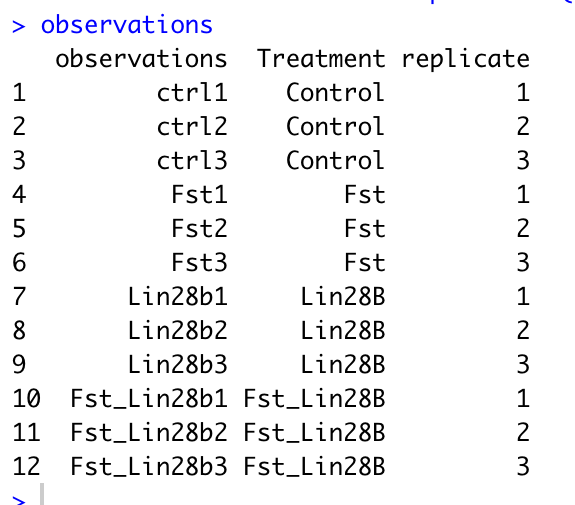
names(obs) = c("observations","genotype", "gender")Below is an example observation file.
- The first column must be named observations with rows corresponding to your sample names.
- You can add any additional columns that you would like to be able to use for comparison in gEAR.
- If you have replicate samples, to enable all the functionalities of the statistical tools in gEAR, please include a column with the name 'replicate' and values similar to below.
- A column named "replicate", “BioRep”, or “TechRep” is needed for conducting statistic analysis with the comparison tool. The code for adding a replicate column is below, just change the names in the group_by function to match your own column names.
# Adding a replicate column to your dataframe
# Change the variables (e.g. genotype, gender)in group_by to those in your own dataframe.
obs <- obs %>% group_by(genotype, gender) %>% mutate(replicate = row_number())
obs = data.frame(obs)
write.table(obs, file= "observation.tab", sep="\t", row.names=F, quote=F)- annotation. check whether there is ensemble in the count file
temp=count[grepl("ENS",count[,1]),]
if (nrow(temp)>0) {
print('ensembl ID are detected')
} else {
print('Error: no ensemble detected')}Here we are using the mouse annotation database, if you wish to use a different database run listDatasets(mart) to get a full list of available annotations.
library(biomaRT)
mart = useMart( 'ensembl' )
datasets <- listDatasets(mart)
mart = useDataset( 'mmusculus_gene_ensembl' , mart = mart )
ensembl = getBM( attributes = c('ensembl_gene_id','external_gene_name') , mart=mart)
names(ensembl)[2] = "gene_symbol"
head(ensembl)
#only keep the first ensemble ID for each gene
ensembl.m=ensembl[!duplicated(ensembl$gene_symbol),]
#merge by gene_symbol or ensembl ID
count.ann = merge(ensembl, count, by.x="gene_symbol", by.y="gene_symbol")
names(count.ann)[c(2,1)] = c("ensembl_ID", "gene_symbol")
write.table(count.ann[-1], file= "expression.tab", sep="\t", quote=F, row.names=F)
genes_to_use=count.ann[c(2,1)]
write.table(genes_to_use,file= "genes.tab", sep="\t", quote=F,row.names=F)if the count file is raw count, basic normalization need to be completed before uploading
count_to_norm=count.ann[-c(1:2)]
row.names(count_to_norm)=count.ann[,1]
cpm=sweep(count_to_norm,2,colSums(count_to_norm),FUN="/")*10^6
##quantile normalization of cpm
quantile_normalisation <- function(df){
df_rank <- apply(df,2,rank,ties.method="min")
df_sorted <- data.frame(apply(df, 2, sort))
df_mean <- apply(df_sorted, 1, mean)
index_to_mean <- function(my_index, my_mean){
return(my_mean[my_index])}
df_final <- apply(df_rank, 2, index_to_mean, my_mean=df_mean)
rownames(df_final) <- rownames(df)
return(df_final)}
nq=quantile_normalisation(data.frame(cpm))
write.table(nq, file=paste(outfile_prefix, "expression.tab", sep = ""), sep="\t", quote=F, row.names=T)Compress files together for uploading
system( 'tar -czvf upload.tar.gz *.tab')