An Error-Prone PCR simulator for use in a Mut-Seq study. Made by Samuel Scandrett.
Mut-Seq was developed by Robins et al. (2013). It is a process that couples random mutagenesis, selection, and the depth of NGS to examine the mutability and essentiality of all residues within a protein. Each nucleotide is mutated thee times, creating a library of all possible combinations of mutations. Mut-Seq is scarce in the literature, and one hypothesised reason for the lack of Mut-Seq experiments is due to the time and resources required to obtain accurate parameters for producing the lowest sized mutant library and 100% coverage.
Through the parallel use of bioinformatics and simulations, data can be provided directly to assist a laboratory.
The simulator requires Python 3.8 to work as it was designed.
Older versions of Python3 can be used at risk of failure.
Packages required for use of the simulator can be found in requirements.txt
Simply running the simulator with no parameters will produce an informative output
python epPCRsim_CLI
---- USAGE ----
epPCR.py -i (input_file) -c (cycles) -r (runs) -e (error_rate) -o (output_file)
---- PARAMETERS ----
-c --cycles <int> The number of cycles
-d --deletion <float> The probability for a deletion mutation (default: 0.12)
-e --error-rate <float> The error rate as decimal
-i --input <str> The file to have epPCR performed on
-l --log-output-file Enable log file production containing each mutation and percentage coverage
-n --insertion <float> The probability for an insertion mutation (default: 0.06)
-o --output <str> The name of the output file in fasta format
-r --runs <int> The number of runs (template molecules)
-s --snv <float> The probability for an SNV mutation (default: 0.82)
-x --no-figures Disable figure production
-y --no-coverage Disable production of coverage graph (FASTER)
The simulator seeks to replicate the procedure as if it were to be carried out in a laboratory using cycles that would exponentially increase the number of DNA molecules (Figure 1). This way, the simulated data could be used to support laboratory decisions. For a Mut-Seq experiment, it is important to know details such as how many cycles are required to have one mutation be the most abundant per mutant sequences and what library size is required for 100% coverage.
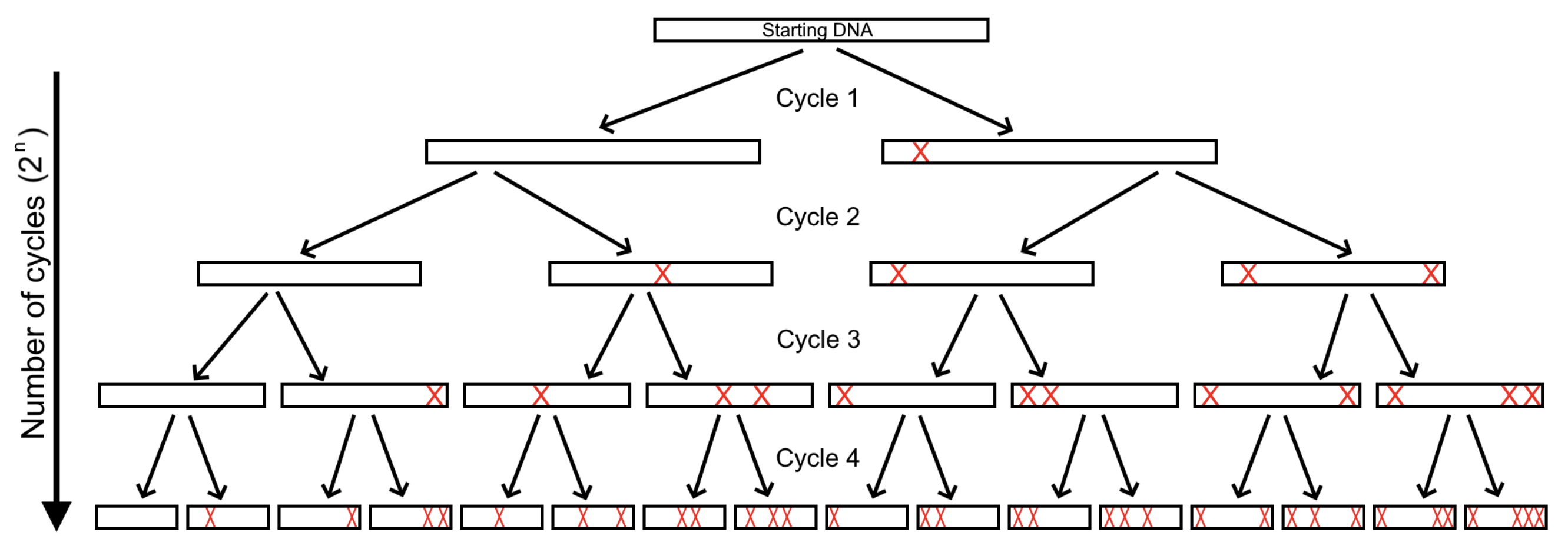 Figure 1. The Error-prone PCR model shares similarity with semiconservative replication, as one strand will always be from the previous
DNA, hence why the left hand side of the model remains unmutated throughout and the right appears the most mutated. Only the single strands
are of importance for the simulation, therefore the DNA rectangles in the model represent a single strand of DNA. For simplicity, the model
assumes a new mutation will occur every cycle. Although this may be unlikely in a practical sense, it visually demonstrates what is occurring
during the process. Identical to PCR, the number of products is 2n where n is the number of cycles.
Figure 1. The Error-prone PCR model shares similarity with semiconservative replication, as one strand will always be from the previous
DNA, hence why the left hand side of the model remains unmutated throughout and the right appears the most mutated. Only the single strands
are of importance for the simulation, therefore the DNA rectangles in the model represent a single strand of DNA. For simplicity, the model
assumes a new mutation will occur every cycle. Although this may be unlikely in a practical sense, it visually demonstrates what is occurring
during the process. Identical to PCR, the number of products is 2n where n is the number of cycles.
One aspect of the real process of epPCR that was unable to be replicated in the simulation was the true number of molecules. The total number of mutant DNA produced following a PCR is:
Number of products = (ng × (6.022 × 1023)) / (length × (1 × 109) × 650)
Instead, the user specified ‘runs’ variable represents the number of molecules that will be simulated undergoing epPCR, therefore, the minimum number of molecules required for 100% coverage could be determined.
The aim of a Mut-Seq experiment is to generate a bank of mutants that collectively cover all three possible nucleotide variants at every position of the gene. For a successful Mut-Seq assay, the desired mutant products should comprise only one mutation in their sequence to investigate phenotypes of single base mutations. Mutants that contain >1 mutations are unusable for the Mut-Seq bank, yet they may still be useful for verifying the effects of deleterious mutations. For generating the bank of mutants, a sequence that contains 0 mutations is more valuable than one that contains >1.
The process for performing a Mut-Seq assay follows the steps in Figure 2. Initially an error rate needs to be defined. Then the cycle number should be set to a cycle number where the number of mutations per mutant equal 1. Finally, a runs number can be determined to scale to 100% coverage.
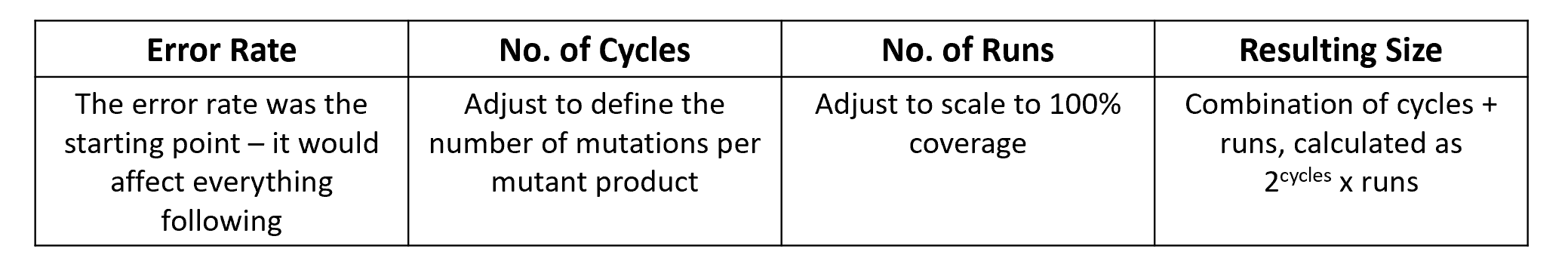 Figure 2. The process for a Mut-Seq assay and achieving 100% coverage.
Figure 2. The process for a Mut-Seq assay and achieving 100% coverage.